今年9月份的药品集采中,降脂药物阿托伐他汀竞争激烈。未中标药企就地解散销售队伍的消息震动业界。
试想,如果有一天,病人只需要每半年打一针,既不需要吃药,也不需要节食,就能维持正常的体重和血脂。到那一天,现有的降脂药物可能被淘汰,那又将是怎样一番景象。随着RNA药物Inclisiran三期临床的成功,这一天也许就在眼前了。
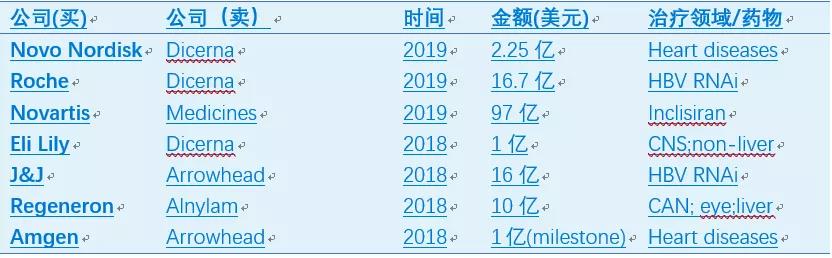
Inclisiran是由Alnylam开发后转让给The Medicines Company的一款靶向PCSK9的小干扰核酸(siRNA)药物。PCSK9是人体变异自证的降脂靶点,已有两款单抗药物批准上市。单抗药物主要是通过抑制PCSK9蛋白与肝脏中低密度脂蛋白受体(LDLR)的结合发挥降脂作用,需要每月注射;而Inclisiran直接降解PCSK9的信使RNA,抑制PCSK9蛋白合成,只需每半年注射一次。多个临床试验表明,Inclisiran降脂效果优于单抗,且副作用与安慰剂相当(1)。其疗效和安全性如果在大规模人群中得到验证,极有可能带来降脂的革命性疗法,甚至实现在健康人群中模拟PCSK9变异,预防与高血脂相关的慢性疾病。正是如此,诺华先下手为强,已于11月25日宣布97亿美元并购The Medicines Company,将Inclisiran纳入囊中。
早在2018年8月,FDA就批准了第一个siRNA药物Patisiran,用于治疗神经退行性疾病hATTR变性;2019年11月20日,又批准了用于治疗急性肝卟啉症(AHP)的第二个siRNA药物Givosiran。他们都是由Alnylam的siRNA技术平台研发成功的罕见病药物。使用脂质纳米颗粒(LNP)技术递送的Patisiran副作用比较明显,注射前需要使用抗组胺药物和激素预防过敏反应。Givosiran和Inclisiran使用的则是更加安全的GalNAc偶联技术。
Inclisiran如果最终获批,标志着siRNA药物进入对安全性要求高,但市场更大的慢病领域。命运多舛的RNA药物将真正迎来“王者归来”的高光时刻。
RNA很长一段时间被认为只是在基因与蛋白质之间传递信息的分子。其实在生命起源之初,RNA才是唯一的生命分子,既可储存信息,也具有酶的功能。亿万年的进化和演变,才产生了DNA和蛋白分子(2)。毫不奇怪,RNA除了充当蛋白合成的信使(mRNA)外,还具有非常重要的调控功能,这样的非转录RNA包括miRNA, siRNA,lncRNA,piwiRNA等等。其中,仅miRNA分子就有400多种,调控至少三分之一的人类基因。自1953年Watson-Crick发现DNA双螺旋结构,人们就产生了根据核酸包含的遗传信息和碱基配对规律实现“程序化”制药的梦想。
在追求这个梦想过程中,RNA药物起步最早,种类包括反义核酸(ASO),小干扰核酸(siRNA),适配体(aptamer),核内小核酸(snRNA),信使RNA(mRNA)等5种。最近有人将基因编辑工具比如GRISPR-Cas9包含的RNA分子也归于RNA药物之列。这样,RNA药物就可以全面覆盖DNA,RNA和蛋白质三大靶点类型。U1snRNA和基因编辑处于非常早期,下面以ASO,siRNA,aptamer和mRNA为重点,简单介绍RNA药物的发展历程。
1978年,哈佛大学科学家Zamecnik等人使用配对的核酸分子抑制RNA病毒的复制,第一次提出了反义核酸(ASO)的概念(3)。
当时的想法比较简单,就是通过碱基配对,封堵DNA或者RNA的翻译和转录。超出人们预期的是,ASO还可以招募RNase酶降解mRNA,干扰pre-mRNA的剪接,甚至还可以间接增强某些蛋白的表达。这些功能都可以被利用,设计针对各种靶点的ASO药物,比如最近获批的药物Nusinersen和Eteplirsen就是通过调控RNA剪接发挥作用。
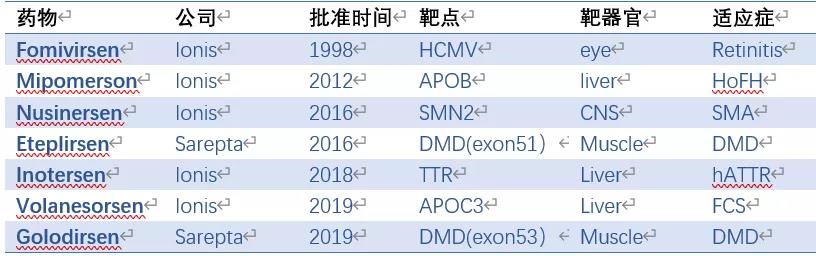
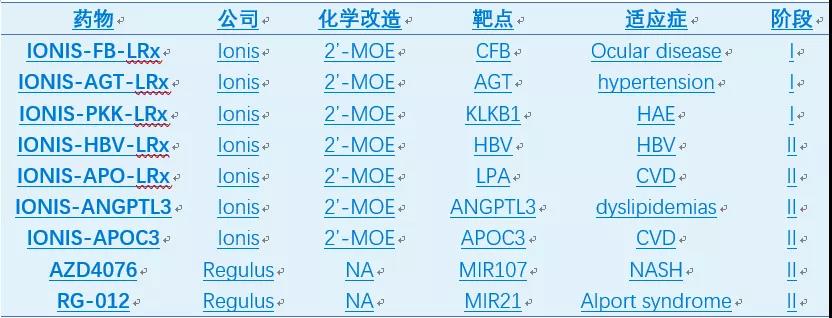
目前已有7款ASO药物被批准上市,两款由Sarepta开发外,其余都来自ASO药物的领先公司Ionis(表1)。
反义核酸药物一路走来,道路并不平坦。1998年,Ionis的第一款药物Fomivirsen批准上市,用于治疗艾滋病人的眼部CMV病毒感染。因为抗HIV药物的快速发展,CMV感染的艾滋病人急剧减少,Fomivirsen最终退出市场。时隔14年,Ionis推出治疗家族性高血脂的APOB反义核酸药物Mipomersen。可惜,由于肝毒性和流感症状,先是被欧洲拒绝批准,后来美国市场也被小分子药物Lomitapide取代,商业上又是一败涂地。2016年,Sarepta开发的Eteplirsen是在DMD患者苦求下获批的,被认为疗效证据不足,许多商业保险机构拒绝报销,市场前景可想而知。
好在Ionis屡败屡战,最近相继推出治疗SMA的药物Nusinersen, hATTR变性药物Inotersen,以及治疗家族性高乳糜微粒血症(FAC)的药物Volanesorsen。其中至少Nusinersen曾经因为疗效太好提前中止临床试验,Inotersen则面临副作用更小的siRNA药物Patisiran的强劲竞争。靶向APOC3的药物Volanesorsen与诺华合作,在心血管领域已进入二期临床,能否与Inclisiran匹敌,让人期待。就在昨天,FDA又批准了Sarepta的第二款DMD药物Golodirsen上市。
当前,全球有超过50个ASO药物处于临床研究阶段,治疗领域覆盖中枢神经系统,心血管,抗感染和肿瘤等。曾经被逼改名的Ionis注定成为未来的巨星,目前管线中拥有5个三期,16个二期,5个一期产品。其中一款治疗糖尿病的二期产品GCGR反义核酸的中国权益转让给了国内小核酸领军企业瑞博生物。
RNA药物真正引起制药界的重视,是因为诺奖成果RNAi的发现。1990年,美国亚利桑那大学的植物学家Jorgensen希望培育五颜六色的牵牛花,于是尝试将深紫色的色素基因插入到植物当中。让人意想不到的是,他只得到了不含色素的白花,检查发现色素基因的表达大大降低了。这个现象让人百思不得其解,因此Jorgensen等人提出了一个所谓基因“共抑制”概念。
后来,Ambrose和Kemphues等科学家发现RNA可以抑制基因的表达,即所谓的RNA干扰现象(RNAi)。1998年AndrewFire和CraigMello 揭示了RNAi的工作机制(5)。
RNAi现象其实是一种降解mRNA的后转录基因沉默(PTGS)。双链RNA在胞质中可以被核酸内切酶Dicer切割成多个小片段(21~23 bp,即siRNA)。siRNA再与体内解旋酶等结合形成沉默复合物(RNA-induced silencing complex,RISC)。RISC诱导降解与siRNA中反义链互补结合的mRNA。RNAi现象高度保守,陆续发现于多种真核生物中。之前提到了植物中的基因共抑制也是因为人工转录基因过程中意外产生的siRNA所介导的干扰现象。
RNAi的特异,高效和简便性使得siRNA迅速成为最流行的基因调控工具。2001年,RNAi技术被《科学》杂志评为2001年的十大科学进展之一。2002年斯坦福大学的Mark Kay在Nature上发文证明人工合成的siRNA可以在小鼠上有效敲低目标基因的表达,这让制药界意识到siRNA在临床应用的可能性。2003年,RNAi被美国财富杂志称为价值亿万美金的生物科技突破。这一年,正值DNA双螺旋结构发现50周年,还发生了一件里程碑事件:人类基因组项目宣布完成。后基因组时代和RNAi技术的突破,让人们觉得“程序化”制药的梦想即将实现。
先前成立的,拥有大量专利的siRNA生物科技公司Alnylam和Sirna迅速成为跨国公司眼里的香饽饽。siRNA药物的第一次繁荣在2006年达到高潮:当年的诺贝尔生理或医学奖颁给了发现RNAi的Fire和Mello;默沙东用11亿美金收购了Sirna;武田,罗氏和诺华纷纷与Alnylam签署巨额合同。
可惜好景不长,在随后的几年内siRNA药物由于递送和免疫反应的问题屡遭挫折。2010年,先是诺华和罗氏中止与Alnylam的合作,随后辉瑞和雅培中止了自己的RNA项目。默沙东坚持多年,于2014年将Sirna以低于原收购价20%的价格卖给了Alnylam。这些动作被媒体放大后,业界普遍认为siRNA只能作为研究工具,成药的希望渺茫。
具有讽刺意味的是,大公司放弃这些资产时,正是siRNA技术取得突破并重新崛起的时候。比如2010年加州理工大学的一项研究证明siRNA人体有效,并能够进入肿瘤组织(7)。2013年,Alnylam公司在新英格兰杂志上发表了patisiran安全有效的临床数据(8)。当然也有慧眼,Arrowhead从2011年到2015年借机收购了大量别人放弃的RNA资产。Alnylam和Dicerna等公司也没有放弃,通过裁员等措施度过了最艰难的时期。
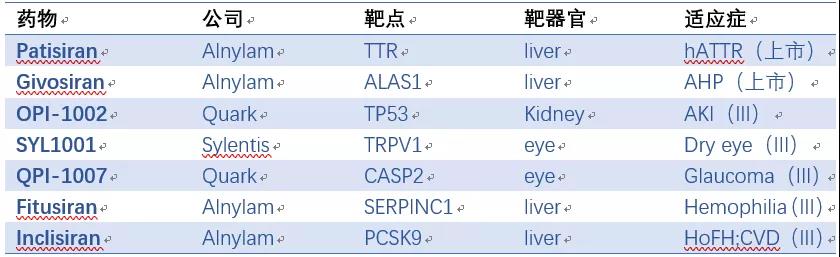
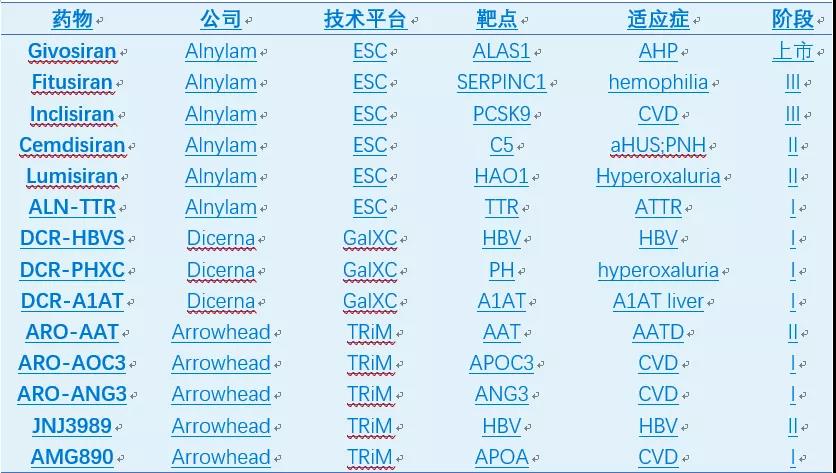
在Alnylam等公司的努力下,siRNA药物目前已有两个上市,至少5个处于三期,9个二期,8个一期(表2)。自2018年起,大公司又开始大肆收购siRNA资产,代表性Deal如表3所示,预示着siRNA药物迎来了第二次浪潮。
在RNA药物里,Aptamer算是一个异类。因为他们不是通过碱基配对来发挥作用,而是类似于蛋白质,依靠自身的三维结构与配体结合。
1990年8月份,美国Colorado大学和麻省总医院的科学家分别在Science和Nature发表文章,显示通过体外进化和筛选,可以得到与目标分子紧密结合的RNA分子,亲合力和特异性与单抗相当(9,10)。
他们把这个筛选过程叫做”Systematic Evolution ofLigands by Exponential Enrichment(SELEX)”。而Aptamer这个名字则是来自拉丁文字“aptus”,意思是“to fix”,中文翻译成适配体。
SELEX符合适者生存的进化原理,RNA文库与靶标孵育一定时间后,未结合的分子被移除,而结合的分子洗脱后经RT-PCR扩增形成新的化合物库,重复上述过程,经过8-20轮筛选,一般能得到高特异性和高亲和力的适配体。这个方法也适应于DNA适配体的筛选。基因测序技术的发展大大加速了适配体的发现,而且这些适配体还可以进一步进行化学修饰,目前学术报道的用于治疗和诊断的适配体超过1000个。
适配体作为药物也有过荣光时刻,2004年FDA批准了第一个适配体药物Pegaptanib,靶向VEGFR,用于治疗老年黄斑病变(AMD)。可惜“既生亮,何生瑜”,那时也正是单抗技术飞速发展的时代。FDA同年批准了治疗肿瘤的VEGFR单抗Avastin,处方外可以用来治疗AMD,疗效和安全性优于Pegaptanib。2006年更优的,治疗AMD的Lucentis上市,Pegaptanib淘汰出局。随后,Fovista(anti-PDGF)和Reg1(anti-FIXa)两款临床阶段的适配体药物相继失败,Reg1甚至造成了一个病人死亡(PEG过敏),适配体逐渐被工业界边缘化。
理论上,适配体免疫原性小,易于合成,靶点范围广,甚至能够结合活性细胞,也能靶向细胞内的靶点,还是有不少独特的优点。然而,随着单抗技术的成熟,大公司已经没有兴趣进行大的投入。有意思的是,长期研究适配体,从学术界转行到工业界的科学家Matthew Levy于2018年发表综述文章,从另一个角度审视了适配体领域衰退的原因(11)。
他认为学术不端造成适配体研究结果不可重复,是药物临床失败和工业界放弃该领域的一个重要原因。Levy试验室用FCS技术重复了大量文献报道的适配体分子,相当一部分根本不可重复。联系文章作者,经常得到的回复是“你们试验做得不对”。指望这种只在自己手里工作的分子将来变成一个药物,Levy认为是在愚弄自己。
Levy指出还有很多试验室专注于表型研究,而不愿意花时间验证适配体本身与靶点的结合是否可靠,而很多时候,观察到的表型是其他原因造成的。更有甚者,为了发文章而罔顾科学事实。比如AS1411是一款结合肿瘤细胞表面抗原nucleolin的抗癌药物。研究表明,AS1411虽然结合nucleolin,但是不能造成nucleolin内吞,但是仍有大量通过AS1411/nucleolin递送药物进入肿瘤细胞的文章发表。Levy呼吁,适配体是好的药物分子,但研究领域需要改变,提高结果的可重复性,让工业界重拾信心。中国科研诚信问题最近成为热点,Levy提到的这些教训尤为应景,值得思考。
1961年,在DNA双螺旋结构发现8年之后,Brenner等人发现DNA和蛋白质之间原来还有“中间人”,即mRNA(12)。没有这个中间人,基因这个生命的蓝图除了一串化学符号之外什么都不是。mRNA的结构,功能和代谢立即成为研究的热点。
1990年,威斯康辛大学科学家首次证明,在动物上直接注射体外转录的mRNA可以表达蛋白(13)。1992年,美国Scripps研究所的一项工作展现了mRNA治疗疾病的潜力,他们通过脑内注射Vasopressin mRNA,成功缓解了大鼠的尿崩症(14)。可惜,在随后的20年内,这是唯一的,mRNA作为蛋白替代疗法的尝试。主要是由于mRNA稳定性和免疫原性问题,人们把注意力更多地放在体外稳定的plasmid DNA上。然而,人们逐渐发现DNA作为药物,问题也很多。比如,药物需要入核,还容易造成基因插入突变等。随着化学改造和药物传递技术的发展,RNA药物的稳定性和免疫原性逐渐得到克服(15),mRNA作为药物的优点和潜力慢慢展现。
首先,mRNA虽然是核酸分子,但与DNA不一样,他们的作用是暂时的,完成工作后即被降解,造成基因突变的风险很小;
第二,一旦靶点确定,mRNA药物的发现和设计几乎是程序化的,无需费时费力;
第三,碱基及其类似物,RNA合成酶和固相合成设备的商业化让mRNA的合成变得简便。生产成本也大为下降。有专家估计,在当前的技术水平下,mRNA药物的生产成本是单抗的十分之一左右。这些特点让mRNA的研发和生产的速度大大加快。2013年,中国爆发禽流感,中国疾控中心公开病毒基因序列后第8天,诺华疫苗部门就完成了一个mRNA疫苗的发现和验证,创造了一个目前还无人打破的纪录(16)。这种速度完全可以与病毒变异保持同步,有望彻底解决棘手的耐药问题。
第四,一个mRNA药物可以同时表达多种蛋白,这为多价疫苗,比如肿瘤个性化疫苗,以及多蛋白联合用药提供了独特的便利性。这个特点甚至可以用来设计自我复制的mRNA药物组合,也就是靶点mRNA加全套复制蛋白的mRNA。受制于递送效率, mRNA分子只有极少(0.01%)能成功进入细胞质并表达蛋白,因此需要大剂量给药,带来副作用。药物自我复制的功能有望帮助降低给药剂量。
第五,ex-vivo细胞治疗的应用。电转等技术使得体外递送mRNA进入细胞变得十分便利。将靶细胞经体外导入mRNA操作后,再回输病人体内,可以避开直接给药的困难。还可以利用mRNA重编程,体外产生用于治疗的iPSC干细胞。最后,只要递送技术允许,mRNA几乎可以在任何组织或细胞,表达任何蛋白,包括基因编辑工具TALEN和Cas9等。这样CRISPR-Cas9就可以变成一种真正意义上的RNA药物组合,包括sgRNA和Cas9mRNA。
mRNA药物理论上可以覆盖包括单抗,细胞治疗和基因编辑在内的所有当前最热门的新技术。有这样的故事,就不难理解为什么2010年成立的,聚焦mRNA技术的Moderna会成为Biotech历史上最大的IPO。截止今年4月份,Moderna累计募资19亿美元,估值已超过50亿美元。对于这样一个没有任何临床数据出来biotech,投资者的厚爱让人瞠目结舌。
但是世界上恐怕没有比生物科技翻脸更快的了,从高峰到低谷,有时就在一夜之间,只需一个病人的意外死亡。mRNA技术目前还处于非常早期。分子量大,以及蛋白翻译机器对于去免疫原性和增加稳定性所必须的化学改造容忍度低等原因,让mRNA药物的发展滞后于小核酸药物ASO和siRNA。
mRNA领域发展得比较顺利的还是疫苗的开发,这有几个原因:1)疫苗所要求的蛋白表达量比较低;2)mRNA的免疫反应一定程度度上有利于疫苗的效果;3)抗原递呈的DC细胞天生善于吸收各种外来分子,有利于药物的吸收;4)ex-vivo免疫细胞治疗积累的经验比较多。蛋白替代疗法发展就相对难一些。除了前面提到的递送和免疫等因素外,还要一个很重要的原因就是很多蛋白只有正确的折叠,切割,修饰和分泌后才能发挥正常的功能,而这些转录后的调节,有时是组织或者细胞类型依赖的。也就是mRNA需要正确地递送到特定的组织或细胞里,目前的技术水平还很难达到。另外,治疗蛋白的表达量要求高,这意味着大剂量给药,制剂辅料以及化学改造后非天然核酸的副作用难以克服。还要,mRNA本身不是治疗分子,需要体内“生产”蛋白后起作用,其药代动力学比单抗类蛋白药物更加复杂,个体差异更大,难以控制(17)。
RNA药物的潜力巨大,但要进入体内却要面临进化了数亿年,大自然设计的防火墙:1)核酸的分子量和负电荷让其不能自由通过生物膜;2)RNA容易被血浆和组织中RNase酶降解,被肝脏和肾脏快速清除和被免疫系统识别;3)进入细胞后“卡”在内吞小体中无法发挥功能。
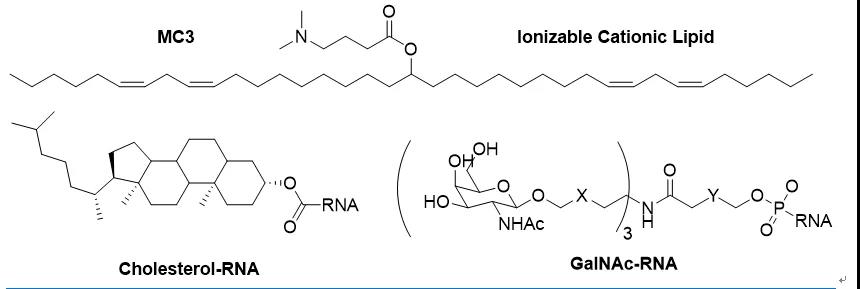
所以半个世纪以来,RNA药物发展面临的技术障碍一直没有变,那就是药物递送。解决递送有两个思路,一个是改造核酸分子,让其稳定并躲避免疫系统的识别;另外一个就是利用药物传输系统,比如说脂质纳米颗粒(LNP)和GalNAc偶联技术。
1)核酸分子的化学改造(Chemical Modification)
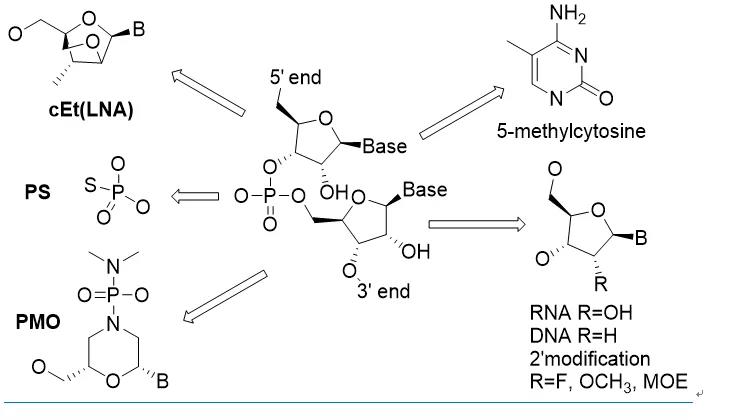
如图1所示,核酸分子的化学改造按部位分三类:碱基,糖环和连接基团磷酸的改造。改造的核酸分子需要考虑多方面的性质,主要包括稳定性,药代性质,碱基配位亲和力,避开免疫系统但确保能够被功能酶所识别,因为ASO, siRNA和mRNA等药物都需要在各种酶(RNase, Ago2 and RNA Polymerase)的作用下才能发挥作用。到目前为止,最具突破性的改造是磷酸连接基团中的一个氧(PO)换成硫(PS)和糖环2位修饰。
德国Max-Planck研究所的科学家在1966年第一次合成报道了PS核酸分子,并意外发现一个原子的改变使得核酸分子可以抵抗酶的降解(18)。虽然稳定性也很重要,但PS对核酸药物最大影响其实是因为硫原子的疏水性增加了药物的血浆蛋白结合,从而大大改善了核酸的药代动力学性质。50多年过去了,虽然有大量的其他尝试,但PS仍然是核酸药物中最常用的磷酸连接基团。
PS提高了稳定性,但却降低了核酸分子碱基配位的亲和力。因为这个原因,第一代ASO药物需要大剂量反复给药,缺点明显,其他改造变得非常必要。这次,老天爷给了些提示。我们知道,RNA分子中糖环的2位是羟基(OH),而DNA的2位没有取代基。早在50年代,人们就发现在自然界存在微量的2位是甲氧基(OCH3)的核酸分子。这提示2位可能可以耐受化学改造。
过去30年,Ionis的化学家在2位改造做了大量工作,为核酸药物的发展做出了不可磨灭的贡献。目前,核酸药物中2位最常见取代包括OCH3,F和MOE,其中MOE表现最为突出。糖环的改造还包括构象锁定类似物(locked nuclearacids, LNA)和吗啉酰胺(PMO)等。这些改造提高了核酸分子的稳定性和亲和力,并可以维持酶识别所需的C3‘-endo构象,以及RNA干扰中RISC识别所需的A-form双螺旋结构。
核酸药物对碱基的改造耐受性较差。ASO和siRNA药物中常见的改造有胞嘧啶的甲基化(5-methylcytosine)。mRNA药物中的尝试相对多一些,包括peudouridine,2-thiouridine, N6-methyladenosine以及各种碱基的5位甲基化。另外,mRNA转录需要5‘-capping,化学改造的cap类似物常用来提高蛋白的转录水平。
RNA药物是多个核酸分子组成的高分子,各种改造的数量和组合对分子的成药性影响很大,比如一般认为需要14个PS才能保证足够的血浆蛋白结合。某个特定修饰可能造成酶识别能力的丢失。因此在药物设计中,需要反复设计测试最合理的组合。有时还需保持一段天然的系列,比如ASO药物中被RNase识别的Gapmer。核酸分子的单体改造与小分子药物不同,如上所述,只有有限的改造空间,而且专利早已过期。因此不同改造的组合,对于IP的产生很重要,这对药物开发至关重要。
2)核酸药物的递送技术(Delivery Tech)
早期核酸的递送主要是病毒载体,以及dextran等非病毒高分子材料。第一代载体的低效和免疫原性等缺点大大限制了核酸药物的发展。
1987年,Felgner等发现带正电荷的脂类分子如DOTMA形成的脂质纳米颗粒(lipid nanoparticle, LNP)能够高效地转染DNA,效率是dextran的5-100倍,被命名为“lipofection”。随后,通过添加辅助的其他类脂物质如DOPE,得到进一步改善的“lipoplexes”。
然而,这些早期的阳离子LNP在体内的应用仍然受到很大限制,比如容易被巨噬细胞清除和产生有害的ROS等。为了解决这些问题,“可离子化“LNP的概念被提出。这些脂质分子在体内保持中性,从而避免被清除和带来副作用,当进入强酸性环境如内吞体,即可以质子化形成阳离子从而与內源的阴离子脂质结合提高跨膜效率。经过筛选,DODAP,DODMA,MC3等一系列可离子化LNPs被开发,从而真正解决了核酸药物的临床应用障碍(图2)。前面提到的第一个上市siRNA药物Patisiran就是使用的MC3-LNP技术。
“可离子化”LNP虽然大大促进了核酸药物的发展,但仍然不尽人意。纳米颗粒的体积比较大,一般需要静脉注射,只能有效进入肝脏,脾脏和肿瘤等空隙较大的组织。另外,LNP的过敏反应也比较严重,在注射之前需要使用抗组胺和激素药物控制。这些缺点让LNP药物只适用于罕见病和癌症等严重疾病。
近年来,ADC等偶联靶向技术在药物传输中得到广泛应用。核酸药物中最先尝试的是脂质偶联(lipidconjugation),比较常见的是与胆固醇偶联。脂质偶联的RNA能够形成类似于低密度脂蛋白(LDL)的聚合物,不但延长了循环时间,而且能与LDL受体或者其他受体结合,通过内吞进入细胞。由于LDL受体在肝脏中高表达,脂质偶联药物系统给药也主要是靶向肝脏,但如局部注射,也能进入皮肤,眼睛和大脑等组织。
让人意外的是,通过偶联技术,RNA药物进入细胞后经常没有功能。研究发现,主要是因为RNA分子通过内吞进入细胞后,被“卡”在内吞小体内,只有约0.01%的分子能够逃逸进入细胞质发挥功能。因此,内吞小体逃逸(endosome escape)成为偶联给药的限速步骤。为了解决这个问题,Arrowhead的科学家做了一个非常有意思的尝试。他们在递送抗乙肝RNAi偶联药物时,同时给予一种从蜜蜂毒素中分离的,可以裂解内吞体膜的多肽melittin,大大提高了RNA分子逃逸的比例。十分遗憾,可能是内吞体的正常功能受到影响,melittin也带来了不可耐受的毒性,Arrowhead停止了三个乙肝项目的临床开发。
RNA偶联药物似乎走进了死胡同。然而,天无绝人之路,一项叫做”GalNAc“的技术横空出世,不但挽救了偶联技术,还成为RNA药物王者归来的真正推手。虽然仅限于肝脏靶点,但GalNAc技术的优势显露无遗:1)只需2-5mg/kg剂量;2)皮下给药;3)一次给药持续6个月以上;4)制剂简单稳定性好。诺华97亿美元收购的降脂药物Inclisiran就是使用的GalNAc技术,安全性和便利性甚至超越了口服小分子。
神奇的发现一般源于意外,GalNAc也不例外。1965年,美国NIH的教授Gilbert Ashwell来到纽约哥伦比亚大学进修。闲暇之时经常去拜访他的好朋友,在Albert Einstein医学院做研究的Anatol Morell教授。Morell当时正在研究糖蛋白ceruloplasmin的代谢,但苦于没有办法测试它的半衰期。做多糖出身的Ashwell建议他用酶剪切水杨酸后,用放射性元素标记糖蛋白末端的乳糖(Glactose)。他们发现这种放射性物质注射到兔子体内,5分钟就从血浆中消失了,几乎100%聚集在肝脏中(20)。
从这开始,他们发现肝脏中存在一种识别乳糖的受体,最后鉴定为哺乳动物中的第一个凝集素蛋白Lectin,并被命名为Ashwell-Morell受体(AMR)。糖蛋白或者表达糖蛋白的细胞暴露末端乳糖即会被AMR回收进入肝脏。每个肝细胞所含AMR超过50万个,这些受体结合乳糖及其类似物后内吞,内吞体中的酸性环境让其释放货物并recycle至细胞表面,这个过程大约只需15分钟。
早在1971年,AMR就被聪明的科学家利用进行靶向肝脏的药物传输(21)。随后,各种乳糖类似物包括亲合力最强的GalNAc被发现可以与AMR结合,并可以用做偶联载体,递送包括小分子,多肽,蛋白和核酸之内的各种药物进入肝脏。1995年,GalNAc被发现可以有效递送核酸分子(22)。GalNAc的数量,linker及连接方式对递送效率的影响得到了广泛的研究。2015年,Alnylam科学家证明GalNAc-siRNA偶联药物可以在小鼠和猴子中抑制抗凝血酶的表达,显示了治疗血友病的潜力(23)。
虽然有扎实的基础研究,GalNAc-RNA药物的临床研究并不是一帆风顺,差点胎死腹中。Alnylam的第一个GalNAc-siRNA临床候选药物Revusiran,靶向hTT基因,用于治疗退行性疾病ATTR。Revusiran二期临床12个月降低血浆TTR水平达90%,却不明原因增加病人的死亡率,导致2016年试验中止。
分析发现,死亡原因可能是这款RNA药物的稳定性不够,需要大剂量给药,而经化学改造的非天然核酸分子大剂量下可能带来毒性。Alnylam随后推出了更加稳定的所谓“ESC“化学骨架(包含2’-OMe/F;PS),成功地解决了这个问题。第二代anti-TTR产品一年的用量只需100mg, 而第一代Revusiran需要28g,剂量降低了280倍,疗效和安全性大为改善。
最近几年,基于GalNAc的技术平台层出不穷,比如Alnylam的ESC,Dicerna的GalXC和Arrowhead的TRiM等。GalNAc-RNA药物已迎来蓬勃发展的时期,至少9个GalNAc-ASO产品处于临床阶段(表5);GalNAc-siRNA发展更为迅速,仅三家公司Alnylam,Dicerna,和Arrowhead产品线即包括一个上市产品(Givosiran),2个三期,4个二期和7个一期产品(表6)。
RNA药物近年来获得了长足的进步,已有7个ASOs,2个siRNA和1个Aptamar上市,超过50个处于临床的各个阶段。Inclisiran明年可能上市,成为第一个进入慢病市场的RNAi药物。取得这样的成绩是过去50年化学,生物和递送技术积累的结果。未来发展方向,可以分为科技和商业二个方面。科技的重点是如何突破肝脏以外的靶器官,这其中的关键是破解内吞体逃逸(endosome escape)的机制,而商业上最大的看点是RNA药物能否带来真正的“单病人”时代。
RNA药物发展的一个最大障碍是靶点局限在肝脏。虽然通过局部注射,在眼睛和中枢神经等其他组织也有成功案例,但疗效和安全性都不尽如人意。继续改造核酸分子的化学骨架,以及优化核酸系列的排列组合,针对不同靶点选择合适的链结构,显然是持续努力的方向,但改善的空间十分有限。
突破肝脏局限最大的希望还在于递送技术的发展,包括改善LNP的设计和结构,国内mRNA领先企业斯微生物的Core-Shell双层递送系统就是一个很好的例子。但终极解决这个问题的关键可能在于破解内吞体逃逸的机制。
前面提到,内吞体逃逸是RNA药物发挥作用的最后一道坎。如果能够提高目前0.01%逃逸率的一个百分点,药物剂量即可大大降低,很多当前困扰RNA药物的问题可以迎刃而解,比如制剂和非天然核酸的副作用,免疫反应等。GalNAc偶联技术就是一个例子。GalNAc在RNA药物上获得巨大成功,除了肝靶向外还大大提高了RNA的逃逸率。不幸的是,我们并不知到为什么。有人猜测是其受体AMR快速 recycle需要高频率打开内吞体膜,给了RNA分子逃逸的机会。在不知道机制的情况下,GalNAc就只能是一个意外,它的成功不可复制。因此,理解生物膜的结构和调控规律,破解内吞体逃逸机制是促进RNA药物发展亟待解决的科学难题。
当然,发现更多肝脏高表达,疾病相关的靶点,短期里也许更切合实际。乙肝和NASH等肝病重症显然是优先发展的领域。深入理解RNA的生物学也是推动领域发展的关键因素,这些RNA既可以是靶点比如致病miRNA和lncRNA等,也可以是药物本身。
另外,促进蛋白表达的,非mRNA药物发展相对滞后,比如ASO,以及华人科学家李龙承教授发现的RNAa等(24)。外源性mRNA有其局限性,比如有时不能被正确折叠和修饰,因此提高內源蛋白表达的RNA“激动剂”有独特的价值。第一个RNAa药物已进入临床研究,通过上调转录因子CEBPA治疗肝癌。相信,随着递送技术的完善,该类药物还有很大的提升空间。
最后,拓展RNA药物的应用范围。基因编辑工具CRISPR-Cas9的核心部件是sgRNA,Rarkau等2019年报道了一种靶向sgRNA的核酸药物,可以抑制CRISPR-Cas9的功能,将来也许可以用作基因编辑的解毒剂(25)。
今年2月份,52岁的美国黑色素瘤晚期病人Brad Kremer遇到了一个“要命”的麻烦。他的一个从德国来的包裹被扣在海关,里面是生物科技公司Bio NTech为他量身定做的肿瘤个性化疫苗BNT122。好事多磨,三个礼拜后他收到包裹并注射了疫苗,随后早已扩散的肿瘤在他身上慢慢消失了。肿瘤个性化疫苗已经带有“单病人”的特点,因为这些疫苗是根据病人所特有的肿瘤新生抗原所设计的mRNA,每个病人的药物独一无二。
“单病人”这个概念是今年10月份新英格兰杂志报道Mila的案例后,FDA两位主任Dr.Woodcock和Dr.Marks在同期杂志上发表评论时提出来的(26)。
2017年,6岁的小女孩Mila被诊断患有罕见疾病“Batten’ disease”,是由于基因MFSD8插入SVA变异造成的。如果没有治疗,女孩很快将会因为大脑退化而死。波士顿儿童医院的Dr.Tiothy Yu为她设计了一款靶向SVA的反义核酸药物,取名”Milasen”,并委托CRO公司完成了定制合成。
通过与FDA的反复沟通,FDA第一次批准了为单个病人研发的药物进入临床研究。Mila注射“Milasen”后,症状得到了控制。FDA两位高管提出了一系列“单病人”时代所面临的监管挑战,比如有效的客观性,安全性的最低要求,批准临床需要什么证据,紧迫性对监管做决定的影响,产品特征的要求,剂量和用药方案,持续生产的问题,以及其他伦理和社会问题。
但一旦开了先河,就很难停止。在病人家属的强烈要求下,Dr.Yu目前正在计划为一名患有Ataxia telangiectasia的婴儿开发反义核酸药物。
“单病人”是个性化药物,或者说是精准医疗的极致,这个时代的开启显然是因为核酸药物的技术发展所推动的。当前技术水平条件下,siRNA药物的一年用药量可以降到100mg以下,给药频率每年二次。mRNA和ASO等药物的设计在计算机辅助下,几乎是分分钟的事情。委托CRO公司生产十分便利,费用也不高。稍微麻烦一点的是制剂的研发和药物的优化测试。随着技术的发展,这些环节也极有可能模块化。
大数据,基因测序,人工智能和3D打印技术必将进一步推动核酸药物和“单病人”时代的发展。试想一下,将来的一天,你感到不舒服,智能手机抽取一滴血,分析发现你的某个基因或者蛋白不妥,指令发给一台药物设计,合成和组装一体化的3D打印设备,1个小时后你就拿到了你自己的药物,用药后疾病一次治愈,终身不再发作。如果不是人体生物这个黑洞,这个梦想从药物技术上看并不遥远。
几十亿年前,是核酸分子孕育了生命。今天,人类面临诸多重大疾病的威胁,解铃还须系铃人,基因治疗和RNA药物的发展,从深层次上显得非常自然和谐。
下个十年,核酸药物有望成为继小分子和单抗之后的第三大类型药物,中国有没有机会,就看我们的科研,监管,资本,和市场能不能形成一个创新生态。在这个生态下,像Ionis和Alnylam这样的公司,30年如一日,可以屡败屡战,经历几起几落,最后王者归来。
中国创新药面临所谓的医保悬崖,有人说me-too新药难做了,急需转向原始创新和原创技术。大方向没有错,但从RNA药物的发展历程来看,原创更难,风险很大。如果,我们不能接受或者无法承受失败,不要轻言从跟随向原创的转型。




文章评论(0)