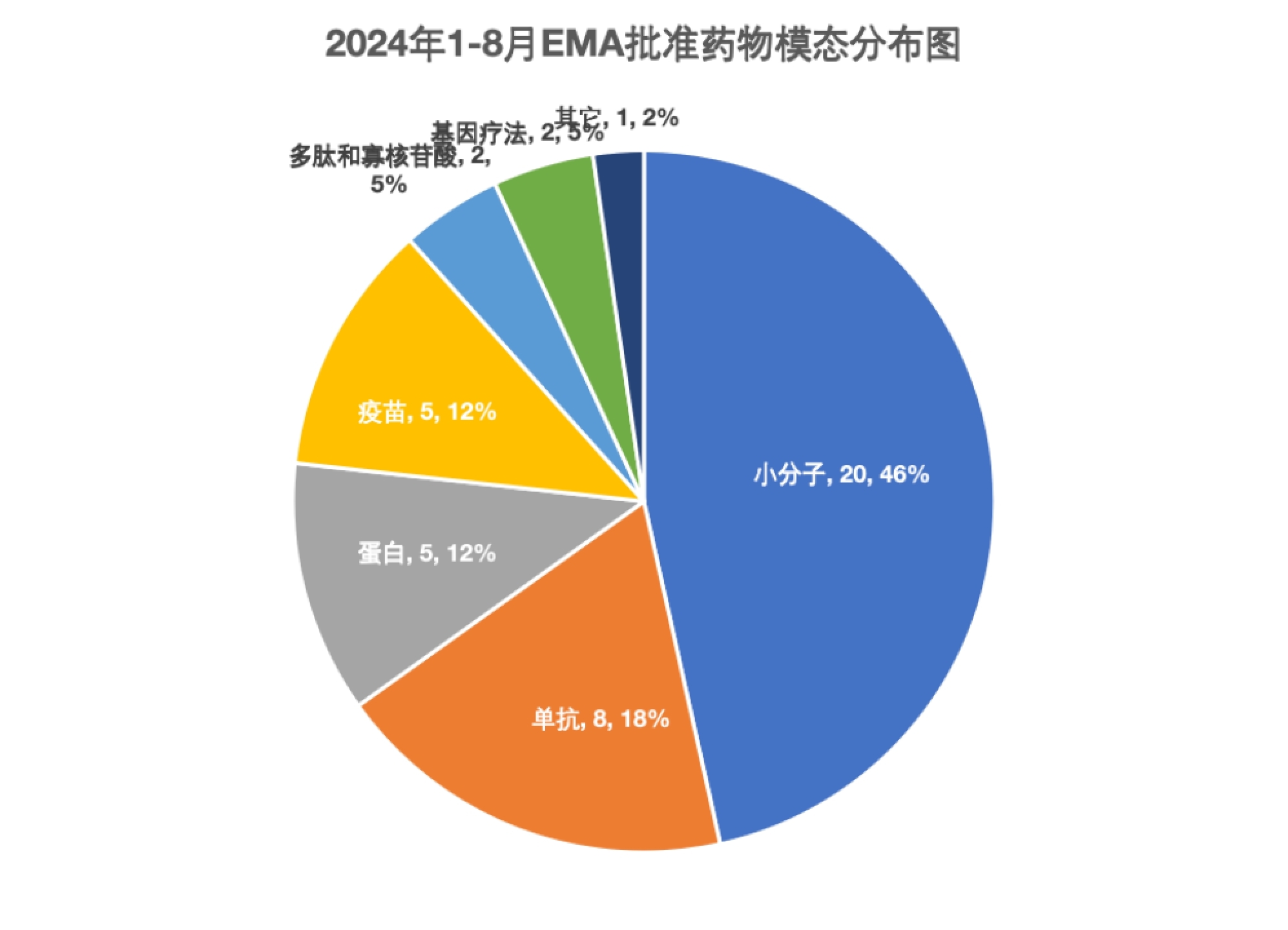
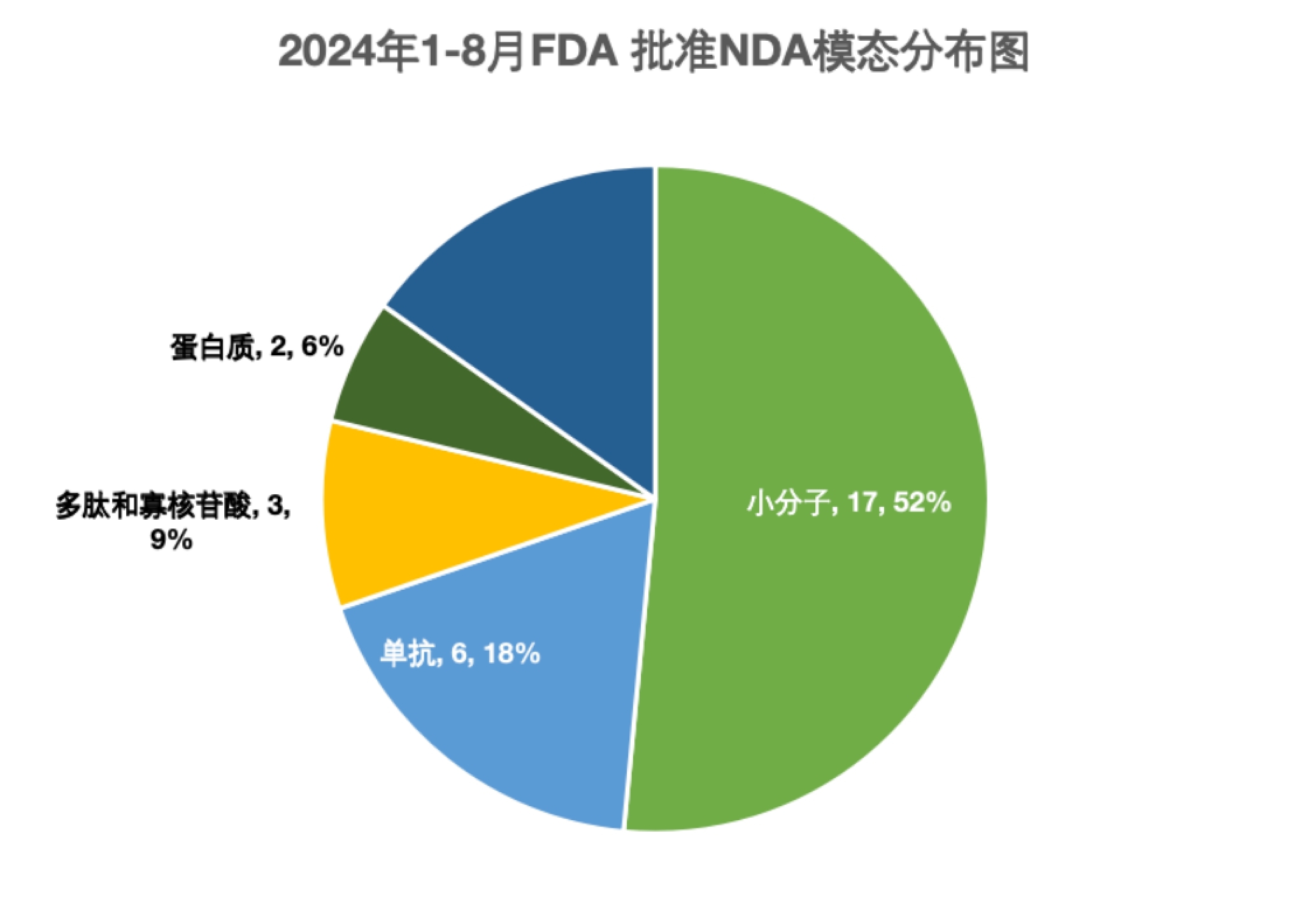
从模态分布来看,EMA批准的药物中小分子药物几乎占据了半壁江山,46%的批准来自小分子。FDA同时期批准的药物中,小分子药物也占据了52%的份额。FDA的CBER今年批准了5款细胞与基因疗法,而EMA只批准了2款。EMA今年批准了5款蛋白产品,FDA目前只批准了2款。
图1. 2024年1至8月EMA批准药物模态分布图。
图2. 2024年1至8月FDA批准新药模态分布图。
欧洲药品管理局在2024年迄今为止只批准了两种CGT法,而FDA已经批准了5款,数字的对比背后可能存在两大监管机构对于CGT疗法的审评标准差别:
CGT疗法的监管环境仍在不断发展,EMA 可能有比 FDA 不同的要求。这可能影响CGT疗法在欧洲的批准数量。EMA 的谨慎态度可能源于CGT疗法的复杂性和潜在风险,需要进行彻底评估以确保安全性和有效性。
FDA 批准的较高数量的CGT疗法可能反映了美国市场对此类疗法开发的更大投资和关注。制药公司可能优先向 FDA 提交申请,因为美国市场规模较大,批准过程可能更快。EMA 的单一批准可能表明在欧洲对基因疗法的接受程度较为缓慢,可能是由于不同的市场动态或监管要求。
公司可能会选择首先寻求 FDA 批准,利用美国市场的规模和 FDA 对创新疗法的既定路径。一旦在美国获得批准,公司可能会随后寻求 EMA 批准,利用美国批准作为支持前例。EMA 的批准过程可能涉及额外的步骤或要求,这可能导致基因疗法的批准比 FDA 更为延迟。
美国的患者可能会比欧洲的患者更早获得创新的基因疗法,这可能导致治疗选择和结果的差异。EMA 的谨慎态度确保只有经过彻底审核的疗法才能进入市场,这对维持高安全标准至关重要,但可能会延迟接触尖端治疗的机会。
EMA 和 FDA 在基因疗法批准数量上的差异突显了监管环境、市场动态和制药公司战略优先级的不同。虽然 FDA 在批准基因疗法方面表现得更为积极,EMA 的更为保守的态度确保了对这些复杂疗法的严格评估。这一趋势突显了理解区域性监管环境及其对患者创新疗法可获得性的影响的重要性。
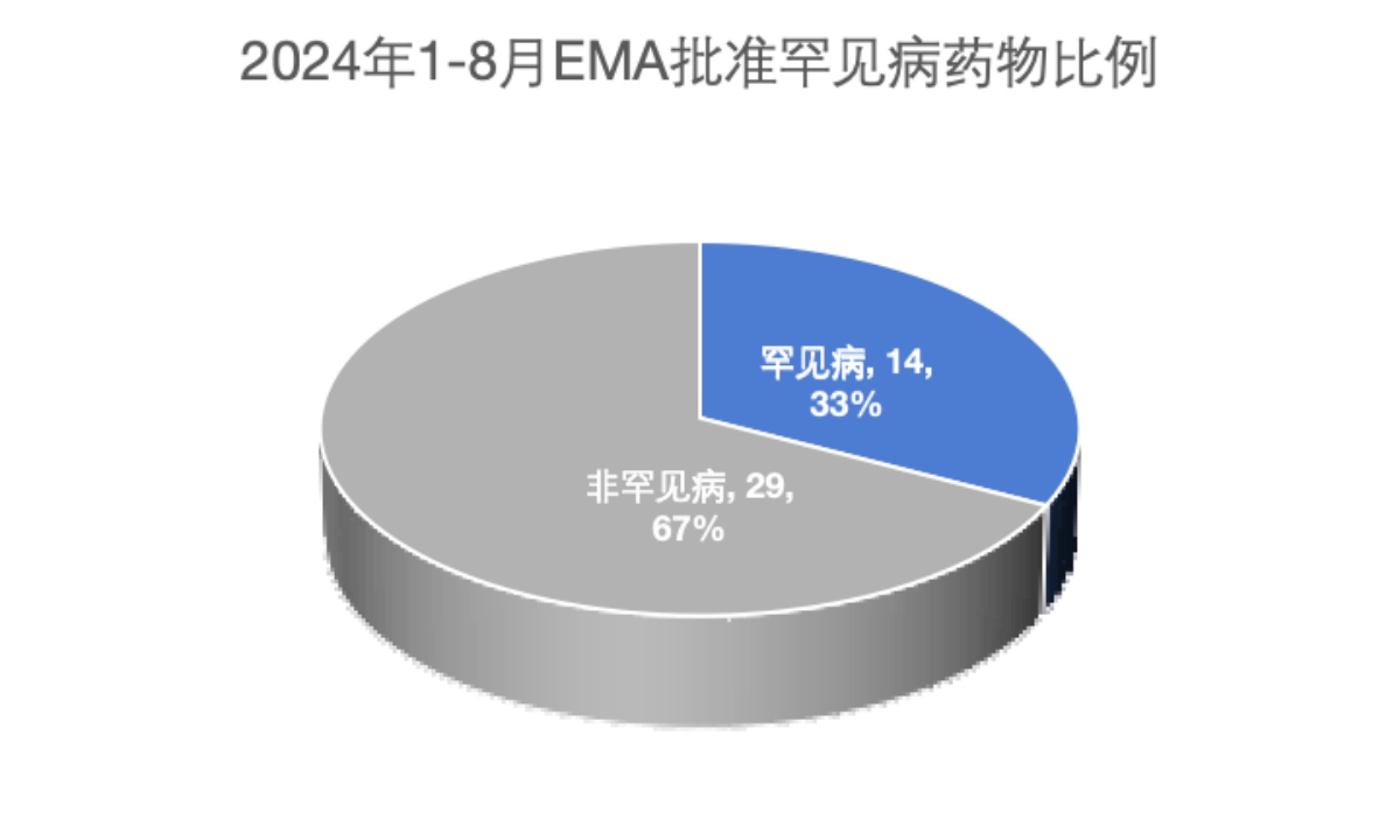
EMA在2024年1至8月批准的药物中有33%针对罕见疾病(图3),这些药物获得了10年的市场独占权,并为中小型生物制药公司提供了额外的激励措施。这表明了监管激励、科学进步和制药行业战略重点在开发罕见疾病治疗中的重要性。
这一趋势对改善患者预后和为罕见疾病患者提供新的希望至关重要。批准针对罕见疾病的药物(也称为孤儿药)表明了EMA在支持开发治疗影响少数人群的疾病的治疗方案方面的决心。这些疾病通常缺乏有效的治疗,开发新的疗法可以显著改善患者的预后和生活质量。
EMA类似于FDA,提供针对孤儿药的激励措施,包括市场独占权、费用减免和科学咨询。这些激励措施鼓励制药公司投资于罕见疾病治疗的研发,否则由于患者数量少,财务上可能不具备可行性。
高比例的罕见疾病批准反映了生物技术和个性化医学的进步。基因疗法、靶向疗法和生物制品等创新正在被开发用于治疗罕见的遗传性疾病和其他罕见疾病。
制药行业越来越注重小众市场,包括罕见疾病,作为其战略优先事项的一部分。这种关注是由高影响力治疗的潜力和对孤儿药开发提供的监管激励驱动的。
对于罕见疾病的患者而言,新疗法的批准可能具有改变生活的意义。这些批准提供了之前几乎没有或完全没有的治疗选择,带来希望,并可能改善生存率和生活质量。
去年, EMA推出了“癌症药物引导者”(Cancer Medicines Pathfinder)计划,以促进癌症药物的开发和审批。该计划着重于加快药物评估,改善与相关方的对话,并有效沟通癌症治疗的益处和风险。
引导者计划专注于新型癌症治疗药物的开发,包括靶向治疗、免疫治疗和联合治疗。通过利用单克隆抗体、检查点抑制剂和基于纳米颗粒的药物输送系统等先进技术,为癌症患者创造更有效和个性化的治疗选择。
该计划促进了领先制药公司、学术机构和研究中心之间的合作。例如,Fulgent Pharma与H. Lee Moffitt癌症中心的合作结合了纳米技术和免疫学的专业知识,以开发靶向癌症治疗药物。
引导者计划支持广泛的临床开发项目,以评估新癌症治疗的安全性和有效性。这包括对非小细胞肺癌、结直肠癌和卵巢癌等多种癌症类型的I期至III期临床试验。
这个计划旨在高效地建立监管环境,确保有前景的新疗法能够及时获得欧洲药品管理局(EMA)和美国食品药品监督管理局(FDA)等机构的批准。这包括为针对罕见癌症的治疗药物获取孤儿药认证,从而加快审批过程。
癌症药物引导者计划的核心在于改善患者预后。通过关注个性化医学和利用基因及基因组检测,引导者计划旨在根据个体患者的特征量身定制治疗方案,从而提高疗效并减少不良反应。
引导计划具有全球视角,临床开发和商业化努力覆盖多个国家,包括美国、中国和各种欧洲国家。这确保了创新癌症治疗药物能够被更广泛的患者群体所接触。
在全球制药批准的复杂背景下,欧盟和美国监管机构之间的差异尤为显著。
例如,Regeneron的Ordspono(odronextamab)在EMA批准用于治疗血癌时, FDA在EMA批准前五个月已拒绝了这一药物。FDA拒绝推荐Ordspono,主要由于对其确认性试验的成熟度存在担忧。EMA则批准了Ordspono用于两种特定类型的血癌,这突显了监管观点的显著差异,特别是在临床试验结果的市场批准准备情况方面。
类似地,诺和诺德的Awiqli(每周一次的胰岛素icodec)旨在改善1型糖尿病患者的血糖控制,但在美国面临了监管障碍。FDA委员会明确投票反对推荐Awiqli,主要因为对比于1型糖尿病成人患者观察到的微小益处,药物带来的低血糖风险显著增加。这些担忧主要基于ONWARDS 6试验的结果,该试验中接受胰岛素icodec治疗的成人患者经历了比接受每日一次胰岛素degludec治疗的患者更高的2级和3级低血糖发生率,这引发了对药物安全性和风险缓解策略充足性的质疑。
此外,尽管Outlook Therapeutics的Lytenava获得了EMA的批准,但在FDA的批准过程中遇到了重大障碍,包括若干CMC问题。预批准制造检查的观察结果以及对进一步确认性临床证据的需求,导致FDA拒绝了这一药物的批准。
导致FDA和EMA在对待同一药物申请时采取不同的监管决定的原因是多重的,可能包含以下几个方面:
FDA有特定的药物审批路径,包括NDA和BLA。FDA还设有加速审批路径,用于满足未满足的医疗需求的药物。
EMA也有类似的路径,但还包括“特殊情况”(Exceptional Circumstances)审批机制,在特定条件下可以基于较少的综合数据批准药物。这一机制在FDA中没有直接对应。EMA的“特殊情况”机制提供了更多的监管灵活性,这一区别使得那些在FDA面前折戟沉沙的药物从EMA获得了生机。
FDA非常重视药物成分的质量、生产过程的可接受性和对良好生产规范(GMP)的遵守情况,因此很多药物因为CMC问题收到了FDA的完整回复函(CRL)而遭到拒绝。虽然EMA同样强调质量和生产标准,但在具体要求和评估方面可能存在差异,导致对药品质量的结论不同。
FDA的科学评估和风险-收益评估可能与EMA不同,这可能源于对临床数据的不同解读或特定监管要求。FDA和EMA可能对临床试验数据的解读不同,导致对药物安全性和有效性的结论不同。例如,FDA可能需要额外的数据或更严格的有效性证据,而EMA可能会在特定条件下接受现有的数据。
根据Citeline旗下的Pink Sheet数据库,在2023年至2024年4月期间,有108种新药获得了FDA或EMA的批准(或者均获批),其中48种获得了双方共同的批准。这48种药物中有37种(77%)首先获得FDA批准,领先EMA批准时间的中位数为5.1个月。只有11种新药(23%)率先获得EMA批准,但领先时间中位数为11.1个月。在重要医学领域,特别是肿瘤学和中枢神经系统治疗领域,FDA更有可能在EMA之前批准药物。
选在首先在FDA还是EMA申请许可,药物开发决策通常是根据各个公司的优先度设定和经济状况指导而决定的,但报销机会和监管可预测性的差异可能导致了FDA在EMA之前批准药物。不仅是相对于EMA,FDA的先发优势在全球范围内也表现得非常明显。
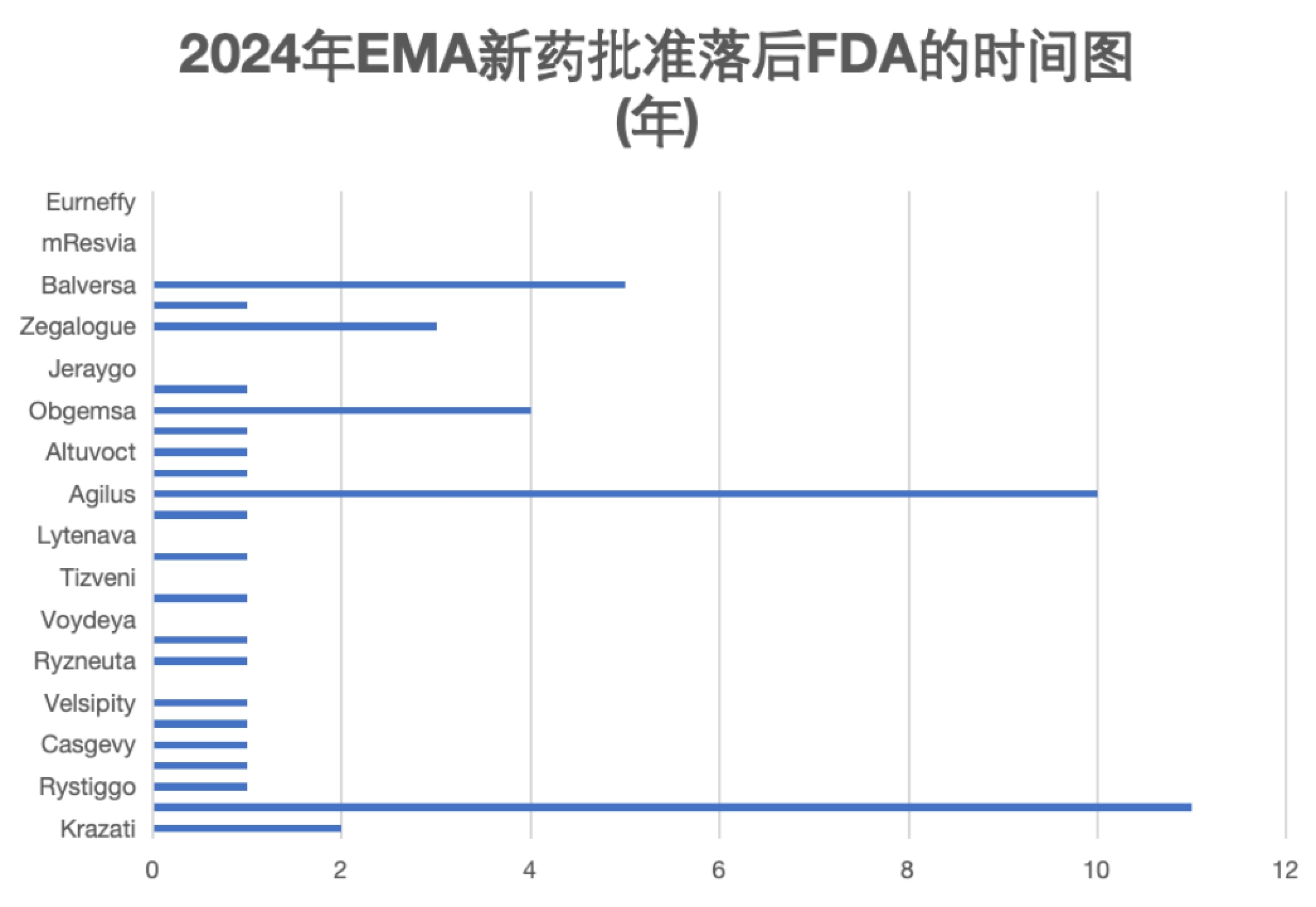
图4. EMA 2024年迄今为止批准新药落后FDA的时间图
2024年1至8月份的药物分析同样印证了以上的发现(图4)。不算EMA今年批准的仿制药,可以发现,EMA今年批准的药物中有31款也获得了FDA的批准,这31款药物中有10款EMA与FDA均在同一年批准,即2024年;15款FDA在一年前的2023年批准;剩下的产品中,FDA领先EMA 2,3,4,5,10,11年批准的药物各有1款。

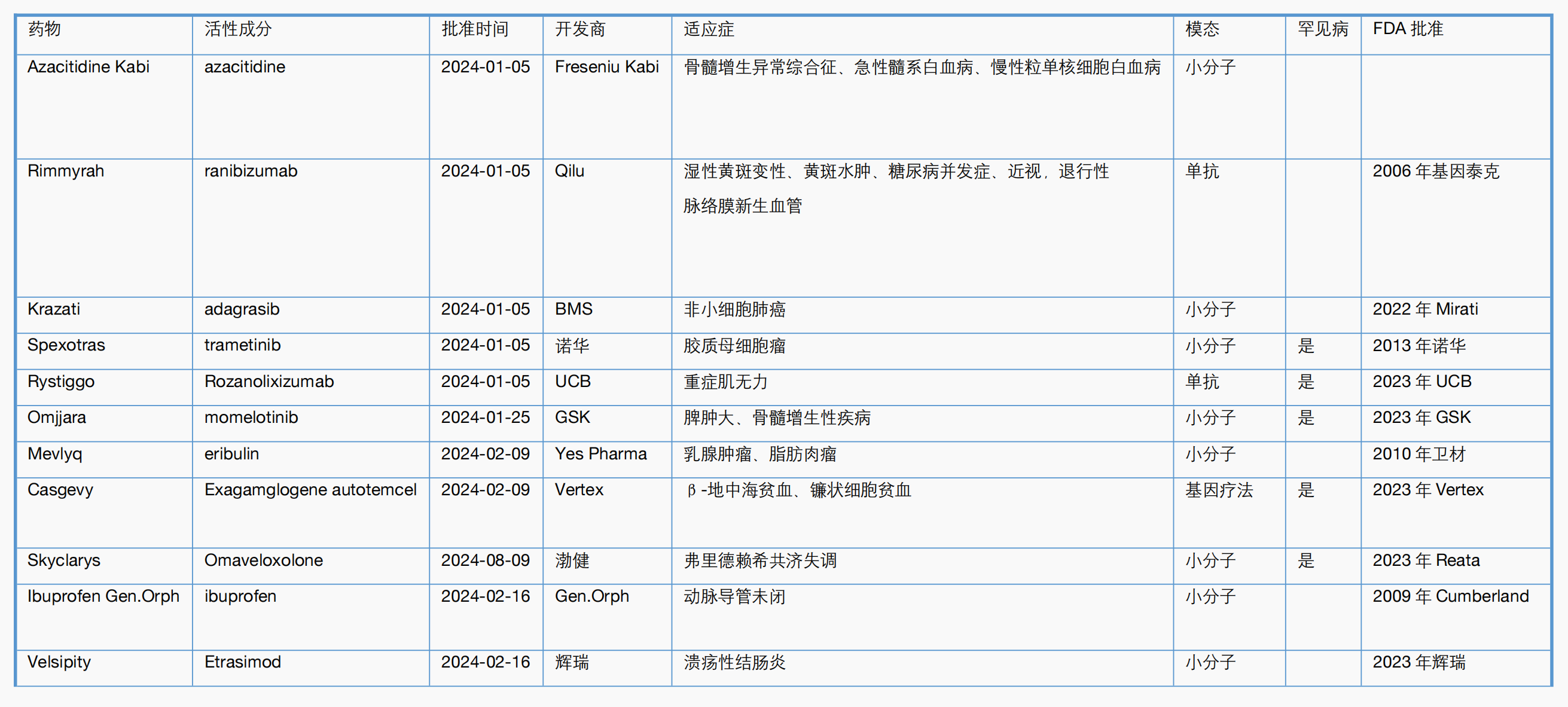
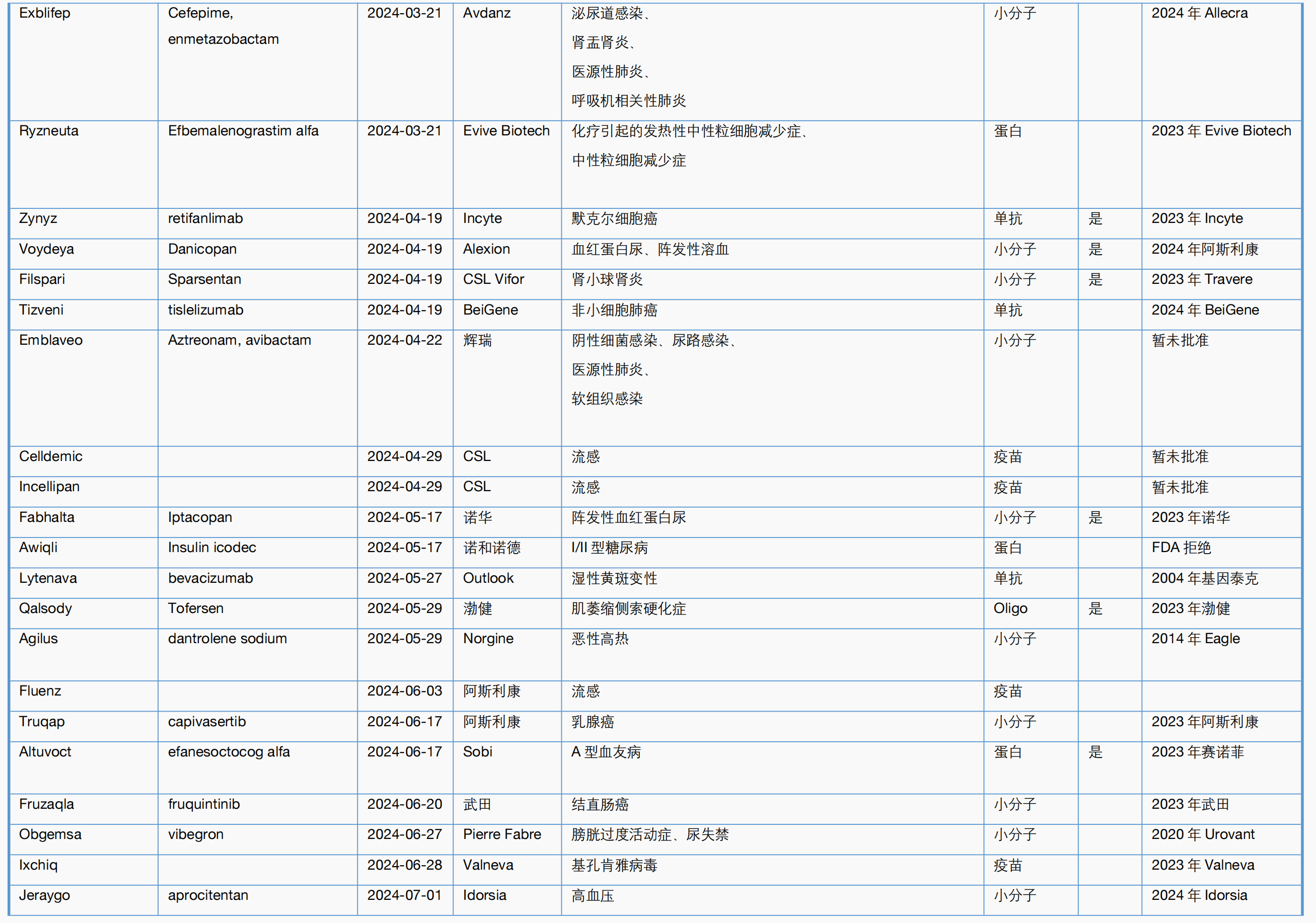
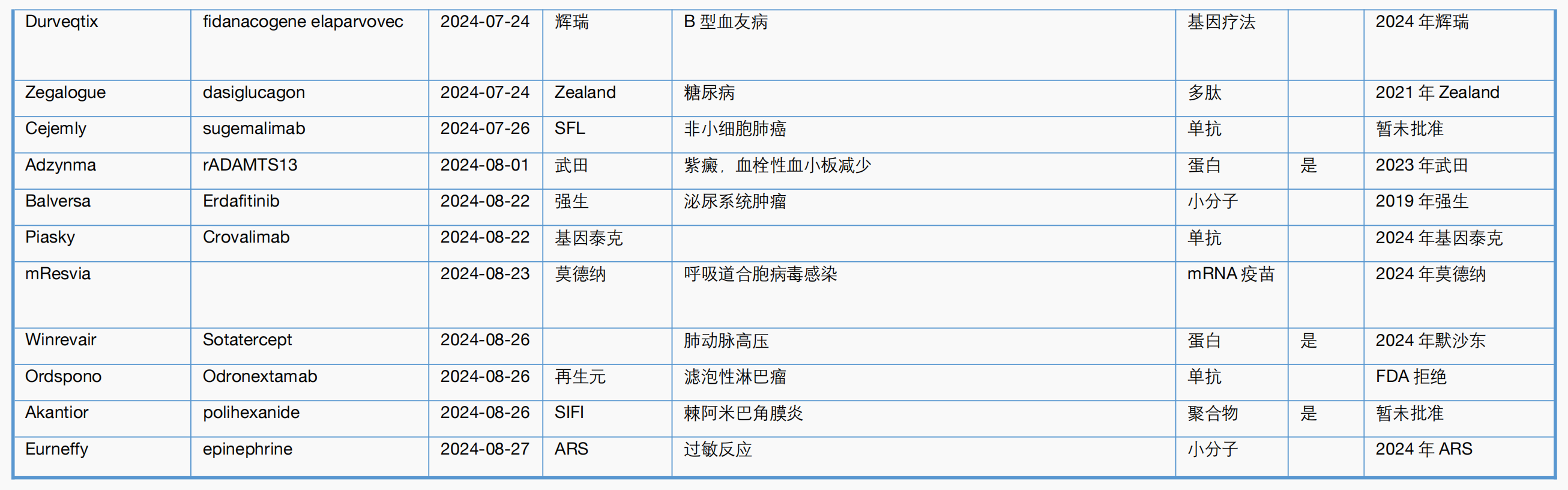
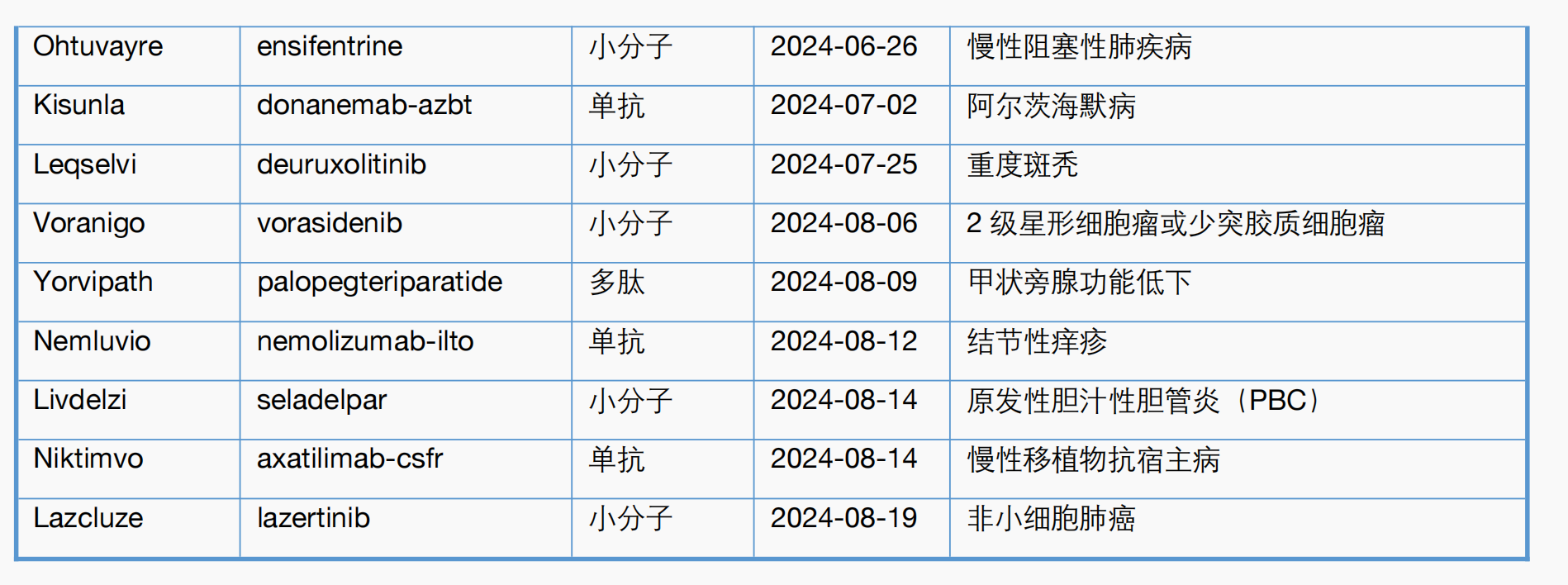
附表1. CDER 2024年一至八月新药审批一览表
附表2. CBER 2024年一至八月主要新药审批一览表
Ref.
New EMA Medicine Approval Through August 2024. Maven Bio. 04. 09. 2024.
Silverman, B. Pharma Looks To America First: US FDA Holds Overwhelming Lead Over EMA In Novel Approvals. Pink Sheet. 05. 07. 2024.



文章评论(0)